Runs vegan::betadisper() on a distance matrix stored in a psExtra
object, usually produced by dist_calc(). This can also be used on the
result of dist_permanova() to ensure that dispersion and PERMANOVA results
correspond to the same distance matrix and sample set.
Arguments
- data
A
psExtraobject containing a distance matrix, as returned bydist_calc()or by downstream functions such asdist_permanova().- variables
Character vector of sample-data variable names to use as grouping variables. Variables must be categorical or coercible groupings; unsupported variable classes are skipped with a warning.
- method
Either
"centroid"or"median". Passed tovegan::betadisper(type = ...).- complete_cases
Logical. If
TRUE, samples with missing values in any of the specifiedvariablesare removed before runningbetadisper(). IfFALSE, the function errors if missing values are present.- verbose
Logical. If
TRUE, prints progress messages.
Value
A psExtra object containing betadisper results. Results are stored
by variable name; each entry contains the fitted betadisper model, its
anova() table, and its TukeyHSD() result.
Details
dist_bdisp() fits one betadisper model per grouping variable. For each
valid grouping variable, it also stores the corresponding anova() and
TukeyHSD() results.
vegan::betadisper() tests whether groups differ in their multivariate
dispersion, i.e. their average distance to a group centroid or spatial median.
This is often used as a companion check when interpreting PERMANOVA results.
microViz currently defaults to method = "centroid", whereas recent versions
of vegan::betadisper() default to type = "median".
When complete_cases = TRUE, samples with missing values in any requested
grouping variable are removed once, before all betadisper models are fitted.
This means all returned models use the same filtered distance matrix.
Examples
library(phyloseq)
library(vegan)
#> Loading required package: permute
data("dietswap", package = "microbiome")
# add some missings to demonstrate automated removal
sample_data(dietswap)$sex[3:6] <- NA
# create a numeric variable to show it will be skipped with a warning
dietswap <- ps_mutate(dietswap, timepoint = as.numeric(timepoint))
# straight to the betadisp
bd1 <- dietswap %>%
tax_agg("Genus") %>%
dist_calc("aitchison") %>%
dist_bdisp(variables = c("sex", "bmi_group", "timepoint")) %>%
bdisp_get()
#> Dropping samples with missings: 4
#> Warning: Variable 'timepoint' is skipped as it cannot be used for grouping (class = 'numeric')
bd1$sex
#> $model
#>
#> Homogeneity of multivariate dispersions
#>
#> Call: vegan::betadisper(d = distMat, group = meta[[V]], type = method)
#>
#> No. of Positive Eigenvalues: 122
#> No. of Negative Eigenvalues: 0
#>
#> Average distance to centroid:
#> female male
#> 9.490 8.494
#>
#> Eigenvalues for PCoA axes:
#> (Showing 8 of 122 eigenvalues)
#> PCoA1 PCoA2 PCoA3 PCoA4 PCoA5 PCoA6 PCoA7 PCoA8
#> 4607.2 1925.4 1297.6 1187.5 945.8 738.8 669.5 529.0
#>
#> $anova
#> Analysis of Variance Table
#>
#> Response: Distances
#> Df Sum Sq Mean Sq F value Pr(>F)
#> Groups 1 53.62 53.622 24.924 1.227e-06 ***
#> Residuals 216 464.70 2.151
#> ---
#> Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
#>
#> $tukeyHSD
#> Tukey multiple comparisons of means
#> 95% family-wise confidence level
#>
#> Fit: aov(formula = distances ~ group, data = df)
#>
#> $group
#> diff lwr upr p adj
#> male-female -0.9961165 -1.389383 -0.6028504 1.2e-06
#>
#>
# quick vegan plotting methods
plot(bd1$sex$model, label.cex = 0.5)
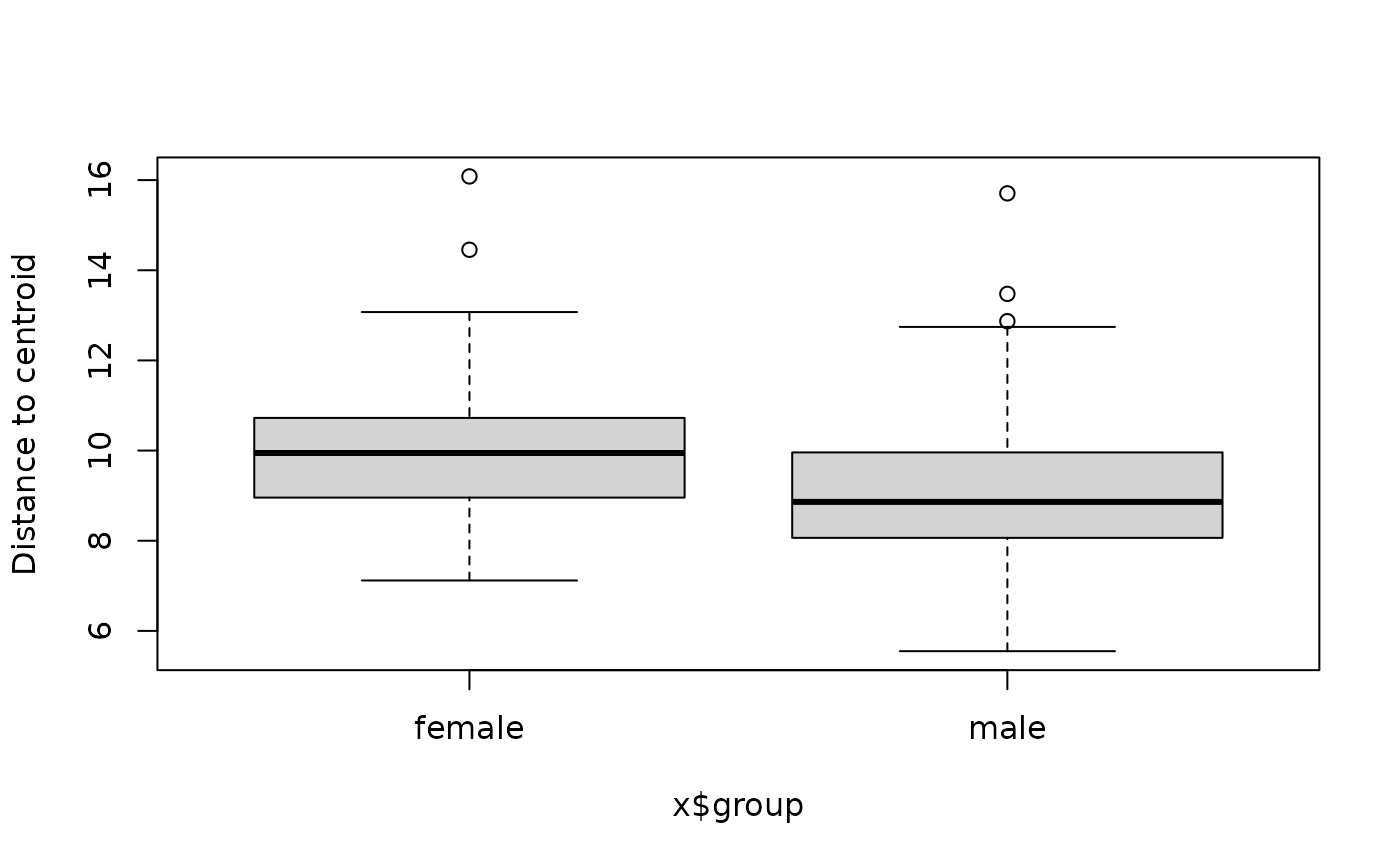
 boxplot(bd1$sex$model)
boxplot(bd1$sex$model)
 # compute distance and use for both permanova and dist_bdisp
testDist <- dietswap %>%
tax_agg("Genus") %>%
dist_calc("bray")
PERM <- testDist %>%
dist_permanova(
variables = c("sex", "bmi_group"),
n_processes = 1, n_perms = 99
)
#> Dropping samples with missings: 4
#> 2026-07-28 13:41:27.92963 - Starting PERMANOVA with 99 perms with 1 processes
#> 2026-07-28 13:41:28.167197 - Finished PERMANOVA
str(PERM, max.level = 1)
#> Formal class 'psExtra' [package "microViz"] with 15 slots
bd <- PERM %>% dist_bdisp(variables = c("sex", "bmi_group"))
bd
#> psExtra object - a phyloseq object with extra slots:
#>
#> phyloseq-class experiment-level object
#> otu_table() OTU Table: [ 130 taxa and 218 samples ]
#> sample_data() Sample Data: [ 218 samples by 8 sample variables ]
#> tax_table() Taxonomy Table: [ 130 taxa by 3 taxonomic ranks ]
#>
#> psExtra info:
#> tax_agg = "Genus"
#>
#> bray distance matrix of size 218
#> 0.7639533 0.731024 0.7283254 0.6637252 0.7437293 ...
#>
#> permanova:
#> Permutation test for adonis under reduced model
#> Marginal effects of terms
#> Permutation: free
#> Number of permutations: 99
#>
#> vegan::adonis2(formula = formula, data = metadata, permutations = n_perms, by = by, parallel = parall)
#> Df SumOfSqs R2 F Pr(>F)
#> sex 1 0.361 0.00933 2.1539 0.17
#> bmi_group 2 2.377 0.06143 7.0888 0.01 **
#> Residual 214 35.874 0.92720
#> Total 217 38.691 1.00000
#> ---
#> Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
#>
#> betadisper:
#> sex bmi_group
# compute distance and use for both permanova and dist_bdisp
testDist <- dietswap %>%
tax_agg("Genus") %>%
dist_calc("bray")
PERM <- testDist %>%
dist_permanova(
variables = c("sex", "bmi_group"),
n_processes = 1, n_perms = 99
)
#> Dropping samples with missings: 4
#> 2026-07-28 13:41:27.92963 - Starting PERMANOVA with 99 perms with 1 processes
#> 2026-07-28 13:41:28.167197 - Finished PERMANOVA
str(PERM, max.level = 1)
#> Formal class 'psExtra' [package "microViz"] with 15 slots
bd <- PERM %>% dist_bdisp(variables = c("sex", "bmi_group"))
bd
#> psExtra object - a phyloseq object with extra slots:
#>
#> phyloseq-class experiment-level object
#> otu_table() OTU Table: [ 130 taxa and 218 samples ]
#> sample_data() Sample Data: [ 218 samples by 8 sample variables ]
#> tax_table() Taxonomy Table: [ 130 taxa by 3 taxonomic ranks ]
#>
#> psExtra info:
#> tax_agg = "Genus"
#>
#> bray distance matrix of size 218
#> 0.7639533 0.731024 0.7283254 0.6637252 0.7437293 ...
#>
#> permanova:
#> Permutation test for adonis under reduced model
#> Marginal effects of terms
#> Permutation: free
#> Number of permutations: 99
#>
#> vegan::adonis2(formula = formula, data = metadata, permutations = n_perms, by = by, parallel = parall)
#> Df SumOfSqs R2 F Pr(>F)
#> sex 1 0.361 0.00933 2.1539 0.17
#> bmi_group 2 2.377 0.06143 7.0888 0.01 **
#> Residual 214 35.874 0.92720
#> Total 217 38.691 1.00000
#> ---
#> Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
#>
#> betadisper:
#> sex bmi_group