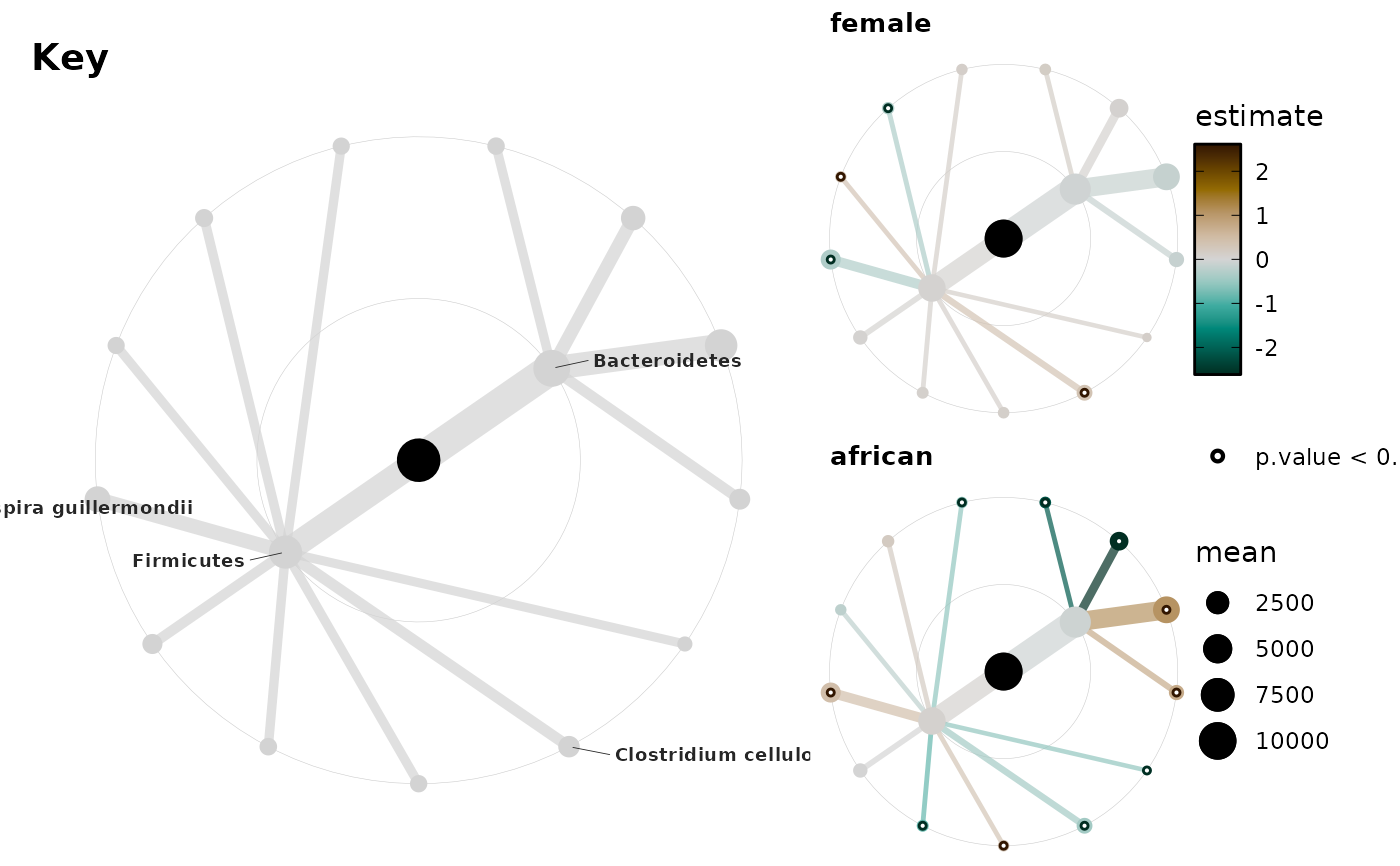
Plot statistical model results for all taxa on a taxonomic tree
Source:R/taxatree_plots.R
taxatree_plots.RdUses a psExtra object to make a tree graph structure from the taxonomic table.
Then adds statistical results stored in "taxatree_stats" of psExtra data
You must use
taxatree_models()first to generate statistical model results.You can adjust p-values with
taxatree_stats_p_adjust()
Usage
taxatree_plots(
data,
colour_stat = "estimate",
palette = "Green-Brown",
reverse_palette = FALSE,
colour_lims = NULL,
colour_oob = scales::oob_squish,
colour_trans = "abs_sqrt",
size_stat = list(prevalence = prev),
node_size_range = c(1, 4),
edge_width_range = node_size_range * 0.8,
size_guide = "legend",
size_trans = "identity",
sig_stat = "p.value",
sig_threshold = 0.05,
sig_shape = "circle filled",
sig_size = 0.75,
sig_stroke = 0.75,
sig_colour = "white",
edge_alpha = 0.7,
vars = "term",
var_renamer = identity,
title_size = 10,
layout = "tree",
layout_seed = NA,
circular = identical(layout, "tree"),
node_sort = NULL,
add_circles = isTRUE(circular),
drop_ranks = TRUE,
l1 = if (palette == "Green-Brown") 10 else NULL,
l2 = if (palette == "Green-Brown") 85 else NULL,
colour_na = "grey35"
)Arguments
- data
psExtra with taxatree_stats, e.g. output of
taxatree_models2stats()- colour_stat
name of variable to scale colour/fill of nodes and edges
- palette
diverging hcl colour palette name from
colorspace::hcl_palettes("diverging")- reverse_palette
reverse direction of colour palette?
- colour_lims
limits of colour and fill scale, NULL will infer lims from range of all data
- colour_oob
scales function to handle colour_stat values outside of colour_lims (default simply squishes "out of bounds" values into the given range)
- colour_trans
name of transformation for colour scale: default is "abs_sqrt", the square-root of absolute values, but you can use the name of any transformer from the
scalespackage, such as "identity" or "exp"- size_stat
named list of length 1, giving function calculated for each taxon, to determine the size of nodes (and edges). Name used as size legend title.
- node_size_range
min and max node sizes, decrease to avoid node overlap
- edge_width_range
min and max edge widths
- size_guide
guide for node sizes, try "none", "legend" or ggplot2::guide_legend()
- size_trans
transformation for size scale you can use (the name of) any transformer from the scales package, such as "identity", "log1p", or "sqrt"
- sig_stat
name of variable indicating statistical significance
- sig_threshold
value of sig_stat variable indicating statistical significance (below this)
- sig_shape
fixed shape for significance marker
- sig_size
fixed size for significance marker
- sig_stroke
fixed stroke width for significance marker
- sig_colour
fixed colour for significance marker (used as fill for filled shapes)
- edge_alpha
fixed alpha value for edges
- vars
name of column indicating terms in models (one plot made per term)
- var_renamer
function to rename variables for plot titles
- title_size
font size of title
- layout
any ggraph layout, default is "tree"
- layout_seed
any numeric, required if a stochastic igraph layout is named
- circular
should the layout be circular?
- node_sort
sort nodes by "increasing" or "decreasing" size? NULL for no sorting. Use
tax_sort()beforetaxatree_plots()for finer control.- add_circles
add faint concentric circles to plot, behind each rank?
- drop_ranks
drop ranks from tree if not included in stats dataframe
- l1
Luminance value at the scale endpoints, NULL for palette's default
- l2
Luminance value at the scale midpoint, NULL for palette's default
- colour_na
colour for NA values in tree. (if unused ranks are not dropped, they will have NA values for colour_stat)
Details
taxatree_plotkey plots same layout as taxatree_plots, but in a fixed colour
See website article for more examples of use: https://david-barnett.github.io/microViz/articles/web-only/modelling-taxa.html
Uses ggraph, see help for main underlying graphing function with ?ggraph::ggraph
It is possible to provide multiple significance markers for multiple thresholds, by passing vectors to the sig_shape, sig_threshold, etc. arguments. It is critically important that the thresholds are provided in decreasing order of severity, e.g. sig_threshold = c(0.001, 0.01, 0.1) and you must provide a shape value for each of them.
See also
taxatree_models() to calculate statistical models for each taxon
taxatree_plotkey() to plot the corresponding labelled key
taxatree_plot_labels() and taxatree_label() to add labels
taxatree_stats_p_adjust() to adjust p-values
Examples
# Limited examples, see website article for more
library(dplyr)
library(ggplot2)
data(dietswap, package = "microbiome")
ps <- dietswap
# create some binary variables for easy visualisation
ps <- ps %>% ps_mutate(
female = if_else(sex == "female", 1, 0, NaN),
african = if_else(nationality == "AFR", 1, 0, NaN)
)
# This example dataset has some taxa with the same name for phylum and family...
# We can fix problems like this with the tax_prepend_ranks function
# (This will always happen with Actinobacteria!)
ps <- tax_prepend_ranks(ps)
# filter out rare taxa
ps <- ps %>% tax_filter(
min_prevalence = 0.5, prev_detection_threshold = 100
)
#> Proportional min_prevalence given: 0.5 --> min 111/222 samples.
# delete the Family rank as we will not use it for this small example
# this is necessary as taxatree_plots can only plot consecutive ranks
ps <- ps %>% tax_mutate(Family = NULL)
# specify variables used for modelling
models <- taxatree_models(
ps = ps, type = corncob::bbdml, ranks = c("Phylum", "Genus"),
formula = ~ female + african, verbose = TRUE
)
#> 2026-07-28 13:42:48.900794 - modelling at rank: Phylum
#> Modelling: P: Bacteroidetes
#> Modelling: P: Firmicutes
#> 2026-07-28 13:42:49.032003 - modelling at rank: Genus
#> Modelling: G: Allistipes et rel.
#> Modelling: G: Bacteroides vulgatus et rel.
#> Modelling: G: Butyrivibrio crossotus et rel.
#> Modelling: G: Clostridium cellulosi et rel.
#> Modelling: G: Clostridium orbiscindens et rel.
#> Modelling: G: Clostridium symbiosum et rel.
#> Modelling: G: Faecalibacterium prausnitzii et rel.
#> Modelling: G: Oscillospira guillermondii et rel.
#> Modelling: G: Prevotella melaninogenica et rel.
#> Modelling: G: Prevotella oralis et rel.
#> Modelling: G: Ruminococcus obeum et rel.
#> Modelling: G: Sporobacter termitidis et rel.
#> Modelling: G: Subdoligranulum variable at rel.
# models list stored as attachment in psExtra
models
#> psExtra object - a phyloseq object with extra slots:
#>
#> phyloseq-class experiment-level object
#> otu_table() OTU Table: [ 13 taxa and 222 samples ]
#> sample_data() Sample Data: [ 222 samples by 10 sample variables ]
#> tax_table() Taxonomy Table: [ 13 taxa by 2 taxonomic ranks ]
#>
#>
#> taxatree_models list:
#> Ranks: Phylum/Genus
# get stats from models
stats <- taxatree_models2stats(models, param = "mu")
stats
#> psExtra object - a phyloseq object with extra slots:
#>
#> phyloseq-class experiment-level object
#> otu_table() OTU Table: [ 13 taxa and 222 samples ]
#> sample_data() Sample Data: [ 222 samples by 10 sample variables ]
#> tax_table() Taxonomy Table: [ 13 taxa by 2 taxonomic ranks ]
#>
#>
#> taxatree_stats dataframe:
#> 15 taxa at 2 ranks: Phylum, Genus
#> 2 terms: female, african
plots <- taxatree_plots(
data = stats, colour_trans = "identity",
size_stat = list(mean = mean),
size_guide = "legend", node_size_range = c(1, 6)
)
# if you change the size_stat for the plots, do the same for the key!!
key <- taxatree_plotkey(
data = stats,
rank == "Phylum" | p.value < 0.05, # labelling criteria
.combine_label = all, # label only taxa where criteria met for both plots
size_stat = list(mean = mean),
node_size_range = c(2, 7), size_guide = "none",
taxon_renamer = function(x) {
stringr::str_remove_all(x, "[PG]: | [ae]t rel.")
}
)
# cowplot is powerful for arranging trees and key and colourbar legend
legend <- cowplot::get_legend(plots[[1]])
plot_col <- cowplot::plot_grid(
plots[[1]] + theme(legend.position = "none"),
plots[[2]] + theme(legend.position = "none"),
ncol = 1
)
cowplot::plot_grid(key, plot_col, legend, nrow = 1, rel_widths = c(4, 2, 1))